
CLINICAL CASE OF A CHILD WITH CONGENITAL NEVUSES
IN THE EYE FUNDUS
A. Popova
Medical Faculty, Chidren's Eye Consulting Cabinet2, University “Alexandrovska Hospital”, Sofia
Детски очен кабинет2 – Диагностично-консултативен център,
УМБАЛ „Александровска“, София
РЕЗЮМЕ
Представя се случай на дете с два пигментни невуса в очното дъно на едното око. Обсъждат се диагностичните и диференциално-диагностичните възможности, а също и рисковете за увреждане на зрителните функции. При рутинното проследяване на детето не са констатирани данни за други очни или извъночни груби патологични прояви. Отбелязва се тенденция за увеличаване на размерите на единия от невусите.
Динамичното проследяване на децата с вродени пигментни промени в очното дъно е необходимост и гаранция както за ранната и точна диагностика, така също и за своевременна профилактика на евентуални усложнения.
Ключови думи: невус, очно дъно, новородено дете, детска възраст
ABSTRACT
A case of a child with two congenital pigmented nevuses in the fundus of one eye is presented. Diagnostic and differential diagnostics, as well as the risks of impairment of visual functions, are discussed. No evidence of other ocular or general condition findings has been reported. There is a tendency to increase the size of one of the nevuses.
The dynamic tracking of children with congenital pigment changes in the ocular fundus is a necessity and a guarantee both for early and accurate diagnosis as well as for timely prevention of possible complications.
Keywords: nevus, eye fundus, newborn infant, childhood
ВЪВЕДЕНИЕ
Клиничното, етиологичното и морфологичното многообразие на хиперпигментните находки в окото е огромно. Понякога то е причина за диагностични, диференциално-диагностични, лечебни и прогностични затруднения, както за зрителните функции, така също и за общото здраве на пациентите.
Хиперпигментните отложения в очното дъно не са нормална находка. Пигментният невус е често използваният синоним за вродена първична хиперпигментация. Вродените пигментни невуси, особено с малък размер и периферно разположение в очното дъно са честа находка при новородени деца.
ЦЕЛ
Да се представят очните промени при дете с два вродени , невуса в очното дъно на едното око и получените резултати да се обсъдят в диагностичен,диференциално-диагностичен и прогностичен аспект.
МАТЕРИАЛ
Момче, насочено проследявано от офталмолог от новородено в продължение на четири години и шест месеца.
МЕТОД
Приложена е комплексна офталмологична методика за изследването на недоносено дете у нас, която включва скрининг на недоносеното за ретинопатия на недоносеното (РН), обстоен офталмологичен преглед и проследяване във времето, включително с дигитална фундускамера (RETCAM III).
РЕЗУЛТАТИ
От анамнезата на майката и наличната медицинска документация: Дете, родено (2013 г.) по оперативен път, от първа (желана) бременност, с тегло 1700 гр., 37 г.с. В ранния неонатален период при детето са констатирани неонатален омфалит, анемия на недоносеността, интравентрикуларен кръвоизлив в ляво – ІІ-ра степен, интрапартална хипотрофия, майчино-фетална инфекция, аномалия в дясното стъпало (pes equinovarus). В неонатологичното отделение са проведени кислородолечение, антибиотично лечение, една хемотрансфузия. Проведен е своевременно неонатален скрининг за ретинопатия на недоносеността (РН) и периодично наблюдение от офталмолог, включително с дигитална фундускамера (RETCAM ІІІ).
Фамилно детето е обременено по майчина линия със захарен диабет ІІ-ри тип (майка, баба и прадядо). Майката е с гестационен диабет и миопичен астигматизъм на двете очи, с нормална зрителна острота с оптична корекция. Бащата е клинично здрав.
Обективно състояние на 2 месечна възраст:
ДО: Добавъчен апарат и преден очен сегмент – без динамика. Зеница – реагира на светлина. Пречупващите среди са прозрачни. Очно дъно (RETCAM): папила – витална, с ясни граници. В І-ва и ІІ-ра зона – съдове - спокойни с нормален ход и калибър; ІІІ-та зона - в процес на васкуларизация. Макула без рефлекс; ЛО: Добавъчен апарат и преден очен сегмент – без динамика. Зеница – реагира на светлина. Пречупващите среди са прозрачни. Очно дъно (RETCAM): папила – витална, с ясни граници. В І-ва и ІІ-ра зона – съдовете са спокойни с нормален ход и калибър; ІІІ-та зона – в процес на васкуларизация. На на меридиана на 3 ч., темпорално от макулата се вижда невус с рамери 1/12 ПД, а на меридиана на 5 ч. под долната съдова аркада – невус с размери 1/10 ПД.
VOD, VOS – не може да се изследва поради възрастта. Детето има нормален мигателен рефлекс към светлината на офталмоскопа; Нормотонус двустранно.
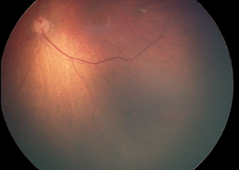
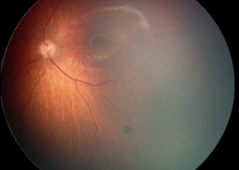
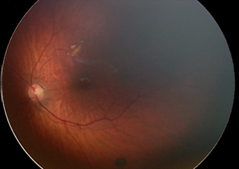
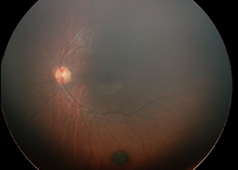
Детето е активно многократно проследявано в продължение на четири години и шест месеца, като не са констатирани прояви за РН (в периода за нейното естествено развитие, а също и по-късно). На една годишна възраст на детето се отчита матуриране на ретиналните съдове, стоп и намаление в размера на невуса на меридиана на 3 ч., но нарастване на диаметъра на другия невус – този на меридиана на 5 ч. в следната последователност: на 2 м. (1/10 ПД); на 5 м. (1/8 ПД); на 9 м. (1/4 ПД), на 1 г. (1 ПД). Същият остава на нивото на ретината и е запазил кръглата си форма и цвят – черен.
Рефракция (на 1 г.): хиперметропия (+3,5 дсф. по вертикала/+4,0 дсф. по хоризонтала), изометропия; Нормотонус; Предписана е сферична корекция – очила (ДО = ЛО: + 2,5 дсфЕ). На Фиг. 1. са представени резултатите от фотодокументацията на пигментната лезия на меридиана на 5 ч., разположен под долната темпорална съдова аркада в лявото око, съответно на 2м. (I.), на 5 м. (II.), на 9 м. (III.) и на 1 година (IV.).
Поредни контролни прегледи (2014 – 2016 г.) – еднакъв резултат с този на 1 годишна възраст по отношение размера, цвета, формата на невусите. Детето расте и се развива нормално. Видимо психомоторното развитие на детето е нормално.
Очен статус (2017 г.): Ортотропия – с и без очилата, във всички погледни посоки. ДО: Клинично здраво; ЛО: Добавъчен апарат и преден очен сегмент – в норма. Прозрачни очни среди. Очно дъно: Хиперпигментацията на меридиана на 3 ч. е на нивото на ретината, точковидна, а тази на меридиана на 5 ч. е също на нивото на ретината, запазила е кръглата си форма, разположение и цвят (черен). Не съществува увеличаване на площта от тази на 1 г. (1 ПД).
Рефракция: хиперметропия (+2,5 дсф. по вертикала/+3,0 дсф. по хоризонтала), изометропия; Нормотонус; Предписана е сферична корекция – очила (ДО = ЛО: + 1,5 дсфЕ).
VOD = VOS = 1,0 с н.к.; Нормотонус двустранно;
ОБСЪЖДАНЕ
В достъпната литература съществува богата информация за хиперпигментните отложения в окото. Характерно за хиперпигментните отложения, не само в окото, но и в човешкия организъм, е тяхното клинично, етиологично и морфологично многообразие, особености, а в някои случаи сходство. Понякога всичко това е причина за диагностични, диференциално-диагностични, лечебни и прогностични затруднения, както за зрителните функции, така също и за общото здраве на пациентите [1 – 29].
Пигментният невус е често използваният синоним за вродена хиперпигментация, независимо от локализацията му в човешкото тяло. Той може да се появи и по-късно след раждането, като в някои случаи е без динамика, в други случаи нарасва, а в други случаи може да се ограничи и видимо да изчезне [2, 12, 25].
Известно е, че хиперпигментните отложения в очното дъно не са нормална находка. Вродените пигментни невуси, особено с малък размер и периферно разположение в очното дъно са честа находка при новородени деца – 1 на 100 новородени при бялата раса и 1 на 10 при кавказките индивиди [12, 21]. Размерите, формата, броя, разположението на невусите в очното дъно и надигнатостта им, както и динамиката, са различни и имат индивидуална характеристика. В периода на естествения бърз растеж на задния очен сегмент (новородено – 3 годишна възраст) е възможно някои невуси да увеличат размера си. Обикновено цветът на невуса е тъмен (кафяв до черен), но образуванието може да бъде с оранжев цвят или да е апигментирано [1, 3 – 9, 12, 14, 26, 27]. Хиперпигментацията може да е с произход от пигментния епител на ретината или от хориоидеята [4, 5, 7, 10, 16 – 21]. В литературата са описани случаи на съчетанието на невусите в очното дъно с други невуси – кожни, конюнктивални, както и с други очни или общи увреждания или усложнения [1 – 3, 6, 7, 13, 15, 16, 23, 25, 26, 28, 29]. Причините за появата на невус в очното дъно са генетични (по-рядко) и/или екзогенни увреждания – в хориоидеята или в пигментния ретинен епител. По-често вродени невуси в очното дъно се констатират при рутинен офталмологичен преглед, в различна възраст на пациента. Зрителните функции са засегнати ако невусът е разположен в макулата или в близост до зрителния нерв, но това са редки случаи [8, 10, 21]. Диференцировката на вродено от придобито хиперпигментно отложение в очното дъно създава затруднения за ранната диагноза и прогнозата за зрителните функции, особено в атипичните случаи, не само при новородени, но и при по-големи деца или възрастни. Все още липсва единно, стандартно становище за класифицирането и субкласифицирането на очните невуси в задния очен сегмент, поради различният им клиничен, етиологичен и морфологичен състав. С малигнена трансформация са до 31 % от пациентите с вроден меланоцитен невус и 1: 5000 с невус на хориоидеята [2, 11, 18, 22, 24].
В разглеждания от нас случай приехме, че най-вероятно се касае за невуси с произход от ретината [Вродена хипертрофия на ретинения пигментен епител - Congenital hypertrophy of the retinal pigment epithelium (CHRPE)], първи подтип.
По литературни данни честотата на CHRPE варира в различните популации в широки граници – от 0,3 % до 40,0 %. Пигментните лезии при CHRPE са с кръгла форма и най-често се констатират случайно, при рутинен офталмологичен преглед на пациента. По-често CHRPE се среща при жени, като съотношението жени към мъже е 2:1.5. CHRPE може да се раздели на три подтипа: 1. Единични вродени пигментни невуси; 2. Групирани CHRPE (тип "мечешки стъпки"); 3. Множествени CHRPE. Подтипове 2 и 3 често се считат за отделни нозологични единици. Обикновено CHRPE нямат клинично значение. Бързият растеж и малигнената трансформация на множествените CHRPE често присъстват при синдрома „Фамилна аденоматозна полипоза“ (FAP – Familial Adenomatous Polyposis), известен в литературата като “Multiple CHRPE” (Nishisho et al., 1991; Rustgi (2007), [15]. FAP е генно детерминирана патология (OMIM#175100) с автозомно-доминантен тип на унаследяване. Клинично първоначално липсват оплаквания. При пациенти, най-често около 25 годишна възраст, с ректално кървене и/или желязодефицитна анемия се констатират полипи (аденоми) в ректума и/или кóлона. Първоначално полипите са доброкачествени, с липса на полова предилекция. FAP е с честота 1,0 % от всички колоректални канцерозни образувания и при 5,0 % от фамилните колоректални канцери. Причина за FAP са мутации в APC (adenomatous polyposis coli) гена, картиран върху дългото рамо на хромозома 5 (5q22.2). APC (611731) е тумор-супресорен протеин . Над 800 мутации в APC гена са причина са FAP. Клинични варианти на FAP са синдромите на Гарднър и Туркот (Gardner syndrome - фамилна колоректална полипоза; Turcot syndrome). При 70,0 - 75,0 % от пациентите с фамилна аденоматозна полипоза се констатират промени в очните дъна от типа на вродена хиперпигментация на ретинения пигментен епител [3, 15, 25, 26]. Нашият пациент не е обременен фамилно в тази насока.
Класическото становище е, че CHRPE не прогресира [4, 9]. Вече е доказано, че при 70,0 – 80,0 % от пациентите с CHRPE съществува нарастване на диаметъра на хиперпигментацията, а в някои случаи нараства и дебелината на лезията. Макар и много рядко, при някои възрастни пациенти, се наблюдава нодулна трансформация в CHRPE, която се дължи на първичен аденокарцином в ретинения пигментен епител [3, 13]. При проучени 330 пациента (на различна възраст) с CHRPE Shields и съавтори констатират среден диаметър на пигментната лезия 4.7 mm (с вариации 0.2-13 mm) и средна дебелина на лезията 0.8 mm (с вариации 0-2.1 mm), като пациентите по-често са с едностранни лезии [22]. Съчетанието на друзи и разнокалибрени пигментни лезии не са характерни за CHRPE и по-често такива находки са суспектни за меланома, най-вече при възрастни пациенти. Приблизително половината от CHRPE лезиите съдържат атрофирани прозоречни дефекти – лакуни. Хистологично лакуните представляват сегментна загуба на ретинения пигментен епител, където вътрешната ретина се слива с мембраната на Bruch. Лакуните варират по форма и размер и са особено видими при по-възрастни пациенти. Хиперпигментните невусни лезии извън макулата и папилата на зрителния нерв обикновено нямат клинично значение [1, 14].
В диференциалната диагноза на вродените невуси в очното дъно е необходимо да се обсъждат вродената хипертрофия на ретинения пигментен епител (като изолирана очна проява или като белег за аденополипозен карцином на ректума. с неговите варианти – при синдромите на Гарднър и Туркот), невус на хориоидеята, меланоза на хориоидеята.
По литературни данни всяка хиперпигментация крие риск за малигнена трансформация в по-късна възраст. Когато невуса показва динамика в размерите, формата или цвета си е препоръчително провеждането на UBM, OCT, FA веднъж годишно. Shields и съавтори описват 40 годишна жена с CHRPE в съчетание с макулна дегенерация, свързана с възрасттта. Подобно описание дават и други автори [1, 20 – 22, 26 ].
Очните критерии за повишен риск за бърз растеж и евентуална малигнизация на невуса са: дебелина на невуса по-висока от 2 mm, оранжева пигментация, близост до зрителния нерв, зрителни смущения, субретинална течност, доказан прогресивен растеж. Според различните автори всеки невус е преканцероза [22].
При наличието на невус в очното дъно или в друга очна структура е необходима оценка за наличието и на кожен невус, независимо от размера му – малък (<1,5 cm), среден (1.5-19.5 см) или голям, гигантски (> 20 см), а също и наличието на други вродени очни или общи увреждания, които крият риск за очното и за общото здраве на пациента [22 – 24, 29]. При нашия пациент липсват други очни или извъночни пигментни находки за наблюдавания период.
Детето остава под периодичен контрол.
ЗАКЛЮЧЕНИЕ
Активното динамично проследяване на индивидите с вродени пигментни промени в очното дъно, особено чрез фотодокументация, подпомага ранната и точна диагностика, а също и своевременната диагностика на евентуалните усложнения.
1. Apte RS, Bressler NM. Foveal congenital hypertrophy of the retinal pigment epithelium in the setting of geographic atrophy from age-related macular degeneration. Am J Ophthalmol. 2003 Jan;135(1):120-1.
2. Bittencourt FV, Marghoob AA, Kopf AW, Koenig KL, Bart RS. Large congenital melanocytic naevi and the risk for development of malignant melanoma and neurocutaneous melanosis. Pediatrics 2000; 106 (4): 736-741.
3. Blair NP, Trempe CL. Hypertrophy of the retinal pigment epithelium associated with Gardner’s syndrome. Am J Ophthalmol 1980;90:661–7.
4. Buettner H. Congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol 1975;79:177–89.
5. Champion R, Daicker BC. Congenital hypertrophy of the pigment epithelium: light microscopic and ultrastructural findings in young children. Retina 1989;9:44–8.
6. Chawla R, Temkar S, Sagar P, Venkatesh P. An unusual case of congenital hypertrophy of retinal pigment epithelium with overlying hemorrhages. Indian J Ophthalmol. 2016 Sep;64(9):672-673. doi: 10.4103/0301-4738.194333.
7. Cohen SY, Quentel G, Guiberteau B, Coscas GJ. Retinal vascular changes in congenital hypertrophy of the retinal pigment epithelium. Ophthalmology. 1993;100:471–4.
8. Fung AT, Pellegrini M, Shields CL. Congenital hypertrophy of the retinal pigment epithelium: enhanced-depth imaging optical coherence tomography in 18 cases. Ophthalmology. 2014 Jan;121(1):251-6.
9. Gass JD. Focal congenital anomalies of the retinal pigment epithelium. Eye 1989;3:1–18.
10. Kaliki S, CL Shields. Choroidal Nevus. AAO, 2015 Focal Points, ISBN 130202015V.
11. Zaal, L. H., Mooi W. J., Smitt, S. J. H., Van der Horst, C M. Classification of congenital melanocytic naevi and malignant transformation: a review of the literature. British Journal of Plastic Surgery 2004; 57: 707-719.
12. Kanski, J., B. Bowling. Clinical Ophthalmology. A Systematic Approach. 7th Edition (2011). Elsevier/Saunders. ISBN-13: 978-0702040931; ISBN-10: 0702040932.
13. Kasner L, Traboulsi EI, Delacruz Z, et al. A histopathologic study of the pigmented fundus lesions in familial adenomatous polyposis. Retina 1992;12:35–42.
14. Lloyd WC 3rd, Eagle RC Jr, Shields JA. Congenital hypertrophy of the retinal pigment epithelium. Electron microscopic and morphometric observations. Ophthalmology 1990;97:1052–60.
15. OMIM, Gardner's syndrome or familial colorectal polyposis (Updated January 26th, 2018).
16. Parker JA, Kalnins VI, Deck JH, et al. Histopathological features of congenital fundus lesions in familial adenomatous polyposis. Can J Ophthalmol 1990;25:159–63.
17. Parsons, M. A., G. Rennie, P. A. Rundle, S. Dhingra, H. Mudhar, A. D. Singh. Congenital hypertrophy of retinal pigment epithelium: a clinico-pathological case report. Br J Ophthalmol. 2005 Jul; 89(7): 920–921.
18. Regillo CD, Eagle RC Jr, Shields JA, et al. Histopathologic findings in congenital grouped pigmentation of the retina. Ophthalmology 1993;100:400–5.
19. Shah SU, Kaliki S, Shields CL et all. Enhanced depth imaging optical coherence tomography of choroidal nevus in 104 cases. Ophthalmology. 2012 May;119(5):1066-72.
20. Shields CL, Mashayekhi A, Materin MA et all. Optical coherence tomography of choroidal nevus in 120 patients. Retina. 2005 Apr-May; 25(3):243-52.
21. Shields CL, Furuta M, Mashayekhi A et all. Clinical spectrum of choroidal nevi based on age at presentation in 3422 consecutive eyes. Ophthalmology. 2008 Mar;115(3):546-552.
22. Shields JA, Eagle RC, Jr, Shields CL, Brown GC, Lally SE. Malignant transformation of congenital hypertrophy of the retinal pigment epithelium. Ophthalmology. 2009;116:2213–6.
23. Schleicher SM, Lim SJM. Congenital nevi, Review. Int J Dermatol 1995; 34: 825-829.
24. Swerdlow AJ, English JSC, Qiao Z. The risk of melanoma in patients with congenital naevi. A cohort study. J Am Acad Dermatol 1995; 32: 595-599.
25. Traboulsi EI, Maumenee IH, Krush AJ, et al. Pigmented ocular fundus lesions in the inherited gastrointestinal polyposis syndromes and in hereditary nonpolyposis colorectal cancer. Ophthalmology 1988;95:964–9.
26. Traboulsi EI, Murphy SF, de la Cruz ZC, et al. A clinicopathologic study of the eyes in familial adenomatous polyposis with extracolonic manifestations (Gardner’s syndrome). Am J Ophthalmol 1990;110:550–61.
27. Wirz K, Lee WR, Coaker T. Progressive changes in congenital hypertrophy of the retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol 1982;219:214–21.
28. Youhnovska P, Toffoli D, Gauthier D. Congenital hypertrophy of the retinal pigment epithelium complicated by a choroidal neovascular membrane. Digit J Ophthalmol. 2013;19:24–7.
29. Zaal, L. H. Giant congenital melanocytic naevi: definition, malignant transformation and treatment modalities, 2009, pp. 121. ISBN: 9789090235486.